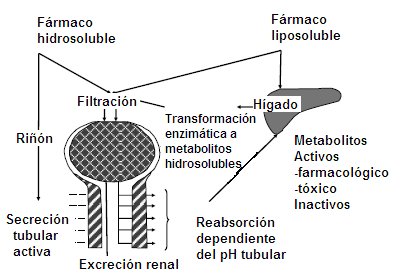
The elimination processes of a drug include two physiological situations: biotransformation and excretion. Biotransformation occurs preferentially in the liver, but not exclusively, since the intestine, placenta and lung can participate in this process, which aims at the enzymatic transformation of any substance exogenous to the body into water-soluble metabolites to facilitate renal excretion. The more fat-soluble a drug is, the longer it will remain in the body. For example, the organophosphate insecticide DDT is so fat soluble that it remains in the liver without being metabolized, so it is not eliminated.
A water-soluble drug can be filtered or secreted in the renal tubule and when it reaches the urine, it is not reabsorbed, so it is eliminated. The enzymatic transformation into water-soluble metabolites can give rise to pharmacologically active metabolites; for example, diazepam has a half-life of 36 hours, but it also has an active metabolite whose half-life is 100 hours, so this metabolite must also be metabolized at the hepatic level for the effect of the drug to cease. When a patient ingests an acute overdose of paracetamol, it destroys hepatic glutathione stores, leading to the production of toxicologically active metabolites that cause acute liver necrosis. The rest of the drugs and their metabolites are inactivated in the liver (Fig. 1).
Figure 1. Drug elimination. Irreversible removal of the drug from the body by all routes. Metabolism is a sequential process that occurs in two phases, involving two enzyme groups.
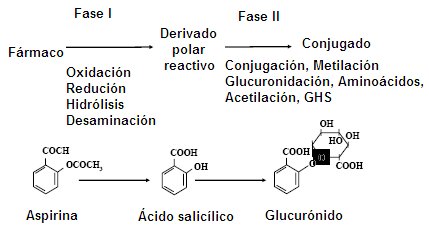
During the phase I enzymatic process, drugs become highly reactive substances; here the hepatic cytochrome P-450 plays a fundamental role. The reactive polar derivative of phase I will be the substrate for phase II enzymes, in which it can undergo glucuronidation, acetylation and methylation processes, in addition to addition of amino acids or glutathione. When the reactive drug is conjugated with one of these molecules, it loses reactivity and liposolubility, that is, it becomes a water-soluble substance that will be easily eliminated. There are drugs that are not metabolized, such as penicillin, which is excreted from the body as it enters. Non-steroidal anti-inflammatory drugs (NSAIDs) are excreted in 98% without alteration and salicylates can undergo some degree of glucuronidation (Fig. 2)
Figure 2. Phases of drug metabolism.
Cytochrome P-450 is a set of proteins with oxidative enzymatic activity that are located in the smooth endoplasmic reticulum (microsomes) of the hepatocyte. This system is nonspecific and easily inducible, that is, its activity increases in the presence of a substance; This is why a smoker patient requires higher amounts of aminophylline than a non-smoker patient, due to the fact that their liver enzymes are induced. In turn, cytochrome P-450 is potentially saturable, because there is a finite amount of enzyme; This means that if an excess of substance is added, the system becomes saturated. It is also easily inhibible; For example, erythromycin is capable of inhibiting liver enzymes that metabolize cisapride, a drug that alone has cardiac toxicity, which is why the cisapride-erythromycin combination should not be used, especially in children, because it can cause fatal arrhythmias The factors that determine the efficiency of the liver to eliminate drugs are: the amount of drug that reaches the liver in a unit of time, which depends on the blood flow and the concentration of the drug in the blood; the concentration of the free drug, that is, that which is not bound to plasma proteins; and the activity of the enzyme systems involved in biotransformation.
Renal drug excretion:
There are three mechanisms for renal drug excretion, which can operate alone or in combination with others: - Glomerular filtration: it is a unidirectional process that depends directly on the free fraction of the drug. Any substance that reaches the glomerulus will be filtered, as long as the molecular size is not too large or that the fraction of the substance that reaches the glomerulus cannot be filtered due to its binding to proteins, because in this way it has a larger molecular size. The normal glomerular filtration rate is 125 to 130 mL / min - Active tubular secretion: requires saturable transporter systems and depends on renal plasma flow (normal value: 425-650 mL / min). In the proximal tubule, these substances can be actively secreted into the tubular lumen, because the affinity of tubular transporters is greater than the affinity of plasma proteins .-- Tubular reabsorption: this process can be active or passive. It is influenced by urinary pH, since non-ionized molecules are fat soluble. If the substance is fat-soluble, it will be reabsorbed in the proximal tubule almost 100%; only those ionized and water-soluble substances will not be reabsorbed and therefore will be excreted.
Clearance:
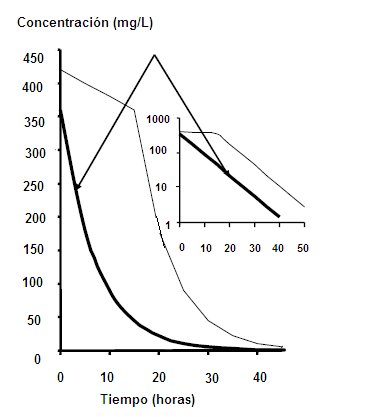
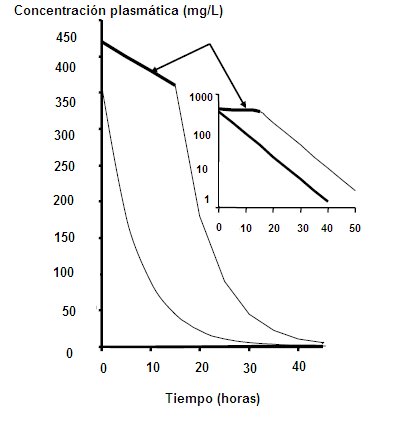
Clearance: Clearance is a descriptive pharmacokinetic parameter; It consists of the analysis of the capacity of the organism to eliminate a drug. Clearence refers to the volume of plasma that is processed, per unit of time, to eliminate a certain drug. The amount of drug removed is proportional to the blood concentration of the drug. If the clearance is very high, it means that the elimination capacity of the organ is enormous; if it is very low, it means that the organ does not have much capacity to eliminate the drug, so that it remains in the body for a longer time, both in the blood and in the tissues. If the drug reaches the tissues, this will influence the amount of drug that exists in the processed volume per unit of time: if there is little drug in that volume, the speed with which it is to be excreted could be altered, because even if it has a metabolic capacity Too large the drug can remain in the body. Therefore, it cannot be estimated that the higher the clearance, the shorter the duration and the lower the clearance, the longer the duration of the drug in the body, which is why the concept of volume of distribution should be introduced to compensate for changes in plasma concentration. Clearance relates the volume of distribution to the mean duration of the drug in the body; the plasma concentration of a drug is a reflection of its volume of distribution (Fig. 3)
Under therapeutic conditions, and considering that an organ is capable of handling a fixed volume of plasma, the percentage of drug that will be eliminated as a function of time is always constant. For example, if there is an amount x / ml of drug in the plasma, in the next unit of time there will be 50% less, that is, 0.5 x / ml and in the next unit of time there will be 25%, or 0, 25 x / ml, that is, a 50% decrease from the above. Therefore, the rate of disappearance of the drug in the body is fast, that is, with kinetics of order 1 (Fig. 4). Figure 4. Kinetics of order 1 When the elimination system is saturated, that is, when there is excess of drug and the system has to handle a fixed amount of drug at a time, in this case the drop in plasma drug concentration is constant, a small amount at a time. If we have a plasma concentration of 500 mg / ml and the metabolic excretion system is saturated, it will only be able to handle a constant amount, for example 15 mg / hour. An hour there will be 485 mg / ml; then, in 2 hours only 30 mg will have come out, when under other conditions 400 would have come out. This corresponds to a kinetics of order 0, where the system is collapsed. This type of kinetics is seen when a patient is intoxicated with Aspirin or ethanol; for this reason, the breathalyzer is extrapolated at the time of the accident, since a small amount is lost, with a linear relationship (Fig. 5). Figure 5. Kinetics of order 0.
Elimination half-life: The elimination half-life is the time it takes for the plasma concentration of a drug to decrease by 50%. When a drug is administered, each half-life there is an accumulation process, but this process is not infinite, but a balance is established between what enters and what leaves, which is called steady state, which produces therapeutic plasma fluctuations in a continuous regime. This state is reached after 4 or 5 half-lives. After 4 half-lives 94% of elimination is completed and 94% of steady state is reached. If a patient with heart failure needs to take digitalis, to reach steady state they would have to receive a daily dose for 7 days; then, a loading dose must be delivered to reach steady state quickly. On the other hand, if digoxin is administered every half-life, in 7 days the steady state is reached, where the amount of the drug entering is equal to that of returning. The elimination half-life depends on the volume of distribution and clearance. If there is a high volume of distribution, it means that there is a lot of drug in the tissues, that is, little drug available to be eliminated and thus the decrease in plasma concentration will be gradual. therefore, if the volume of distribution is high, the half-life of the drug will be longer. The greater the clearance, the shorter the half-life of the drug, because for each volume that the organ processes, all the drug contained in that volume will be eliminated. On the contrary, if the clearance is low, the half-life of the drug will be longer. With these two parameters, dosage regimens can be designed that are useful for drugs such as antiarrhythmic drugs, theophylline, and anticonvulsants, that is, drugs with a narrow therapeutic range. For example, in the case of an arrhythmia, to obtain an immediate effect, a loading dose is given and subsequently, a maintenance dose.The half-life of paracetamol fluctuates between 4 to 6 hours, so to maintain the concentration Plasma should be dosed at appropriate intervals and administered at least every half-life and not only when there is pain. When there is no good therapeutic response to analgesics, it may be because they are being administered at inappropriate intervals. Thus, knowing the half-life of a drug is useful to design, plan and rationalize therapeutic regimens.Conclusions regarding the half-life of a drug: After each dose, the amount of drug in the body increases, the amount of drug eliminated increases. and the rate of accumulation decreases.If the volume of distribution is large, it takes a longer time to reach steady state and excretion is slow.If clearence increases, the time to reach steady state is shorter.If the administration interval of the drug is less than the half-life, the accumulation is toxic.
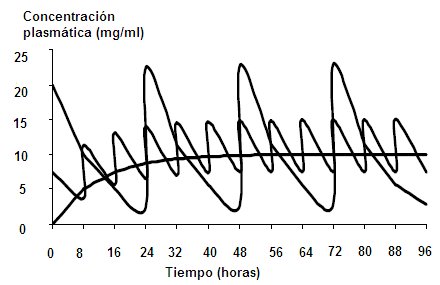
Steady state: When the amount of drug that enters the body is equal to that which leaves, the plasma concentration is said to have reached steady state.A drug accumulates in the body when: the amount that enters the body is greater than that which leaves ; the entry speed is greater than the exit speed; or the same dose is administered at intervals shorter than the half-life. For example, if a patient is given 500 mg of paracetamol every 4 hours, he will have a steady state; but if you do not have a good therapeutic response to the drug and the dose is increased to 1000 mg every 4 hours, the steady state of this new dose of paracetamol will be achieved within 4 half-lives; This occurs when a higher dose is administered in the same time interval, but if this paracetamol is administered every 2 hours, even if it is 500 mg, at 6 hours the patient will be intoxicated with the drug. The interval or the dose can be modified , but there are fluctuations between the maximum dose and the minimum dose. By administering a constant dose of a drug, the dose is adjusted to the mean therapeutic plasma concentration, as is the case with continuous infusion, where the dose is on the average between the maximum dose and the minimum therapeutic dose; but if the drug is administered orally, each half-life there will be a fluctuation in the dose, such that the concentration will decrease by half and then this loss will recover, until it accumulates. If the dose fluctuations are within the therapeutic range, there will be no problem. Sometimes the doses can be modified, increasing them and also increasing the intervals of administration; This can be achieved by knowing the pharmacokinetic parameters of the drug, that is, the volume of distribution, the half-life, the clearance, among others (Fig. 6). Figure 6. Fluctuation of plasma concentrations with respect to the interval and dose of administration. The goals of dosage regimens are: to maintain therapeutic concentrations of a drug at steady state; individualization of therapy; keep plasma concentration within appropriate ranges; it is used preferentially for drugs with a narrow therapeutic window such as anticonvulsants, antiarrhythmics, digoxin and theophylline. Theophylline and carbamazepine are capable of inducing their own metabolism through the enzymatic induction of cytochrome P450, so a dosage regimen should be established considering this fact and the drug doses increased when required. The maintenance dose is the amount The most appropriate drug for administration, in continuous or intermittent infusion, with the aim of maintaining steady state. The calculated value depends on the clearance of the drug; When the administration is orally, the dose will depend on the half-life of the drug and is obtained from the clearence product by the plasma concentration. If the maintenance dose is multiplied by the half-life, the result is the drug delivery interval. The loading dose is the amount of drug needed to quickly reach the desired concentration, within the therapeutic range. It means that the physical reservoirs will be filled with the drug acutely. There is always the risk of reaching toxic plasma concentrations at one time, for this reason digoxin is administered fractionally and not as a bolus. The loading dose depends on the volume of distribution of the drug.
Monitoring of plasma concentrations: It is not done routinely, due to its high cost; furthermore, the risk of toxicity is low and the criteria for monitoring are very strict. It is useful to measure plasma levels in the following circumstances: When there is a clear relationship between the plasma concentration and the desired effect When the plasma concentration cannot be predicted from the drug dose, because there is high variability between patients and little intrapersonal variability When it is difficult to monitor adverse drug reactions (ADRs) or toxic effects When ADRs are similar to the disease, for example lithium levels in bipolar patients, increasing litemia increases the risk of ADRs of the drug. Carbamazepine has a hepatotoxic effect, so it should be monitored.In drugs with a narrow therapeutic margin, such as digoxin or antiarrhythmic drugs.When there is therapeutic failure, that is, lack of adherence to treatment, poor tolerance or poor absorption of the drug.When suspected interaction of another drug that decreases therapeutic effects, for example, the cisapride-erythromycin interaction When overdose or abuse is suspected.


This Medwave work is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported license. This license allows the use, distribution and reproduction of the article in any medium, as long as the corresponding credit is granted to the author of the article and to the medium in which it is published, in this case, Medwave.https: //
www.medwave.cl /link.cgi/Medwave/PuestaDia/Ccursos/3450
